API Usage
{warning} This online page relates to the latest released JChem PostgreSQL Cartridge. Find the version specific documentation in your installed package in
/opt/jchem-psql/doc/directory.
This manual serves as API Developer and User Guide of JChem PostgreSQL Cartridge (JPC). See also Getting started guide for easy setup and use cases.
If you are familiar with JChem Oracle Cartridge (JOC) and are interested in the differences between JOC and JPC, see their comparison here.
CREATE TABLE
Prerequisites
-
jchem-psql service must run
-
extension named chemaxon_type must be created.
CREATE TABLE table_name (structure_column_name MOLECULE('molecule_type_name'));The type of the column where the chemical structures are stored has to be any of the MOLECULE types defined in /etc/chemaxon/types/.
{primary} molecule_type_name must be specified in order to make the column searchable!
Example:
CREATE TABLE ttest (mol MOLECULE('sample'));The created column can be propagated with any of the well-known chemical structural data file formats. Due to postgres type system, the provided string value is automatically converted to MOLECULE type. For importing different molecule formats from local files, see Auxiliary functions for importing into a table.
{primary} Column constraint UNIQUE is not applicable on the MOLECULE type column.
{primary} If you will run updates against a table which contains indexed molecule columns,
to reduce postgresql MVCC index entry creation/deletion traffic, postgres should be able to use HOT.
In order to facilitate the usage of HOT on your tables, use the 'fillfactor' storage argument. Read about fillfactor.
Example:
CREATE TABLE t (m molecule('sample')) WITH (fillfactor=50);
The next chart illustrates how the fillfactor influences the consumed time of the calculation of the molecular weight of 1 M molecules in columns indexed with the domain index - chemindex - applied in JChem PostgreSQL Cartridge.
- Time of the MolWeight calculation in 1M datasets in chemindexed tables created with different fillfactor values *
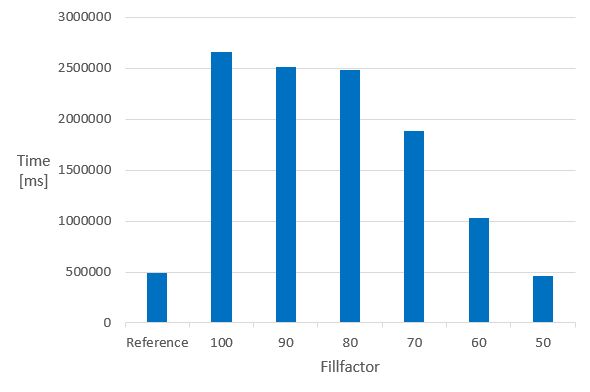
The Reference is the same 1 M dataset, without chemindex, in a table created without fillfactor.
It can be seen that in indexed tables, the speed of the calculation executed in the non-indexed reference table can only be achieved if the fillfactor is set to 50. The 50 % value of the fillfactor means that the memory requirement of the table is approximately doubled compared to the 100 % (or no fillfactor).
Fillfactor 50 is recommended if calculated columns are planned to be added to chemindexed tables.
If no more calculations are planned, the fillfactor can be reset even to 100 by:
ALTER TABLE table_name SET ( fillfactor = 100);
VACUUM FULL table_name;The possibility of dropping the chemindex before such calculations, and creating them after the calculation is always available.
INSERT INTO TABLE
INSERT INTO table_name (column1,column2,...) VALUES (value1,value2,...);Example :
INSERT INTO test (id,mol) VALUES (1,'c1ccccc1'); MOLECULE INDEX
For indexing a column containing chemical structures the following indextypes are provided:
-
chemindex
-
sortedchemindex (available from JPC version 2.6)
You can check whether these indextypes exist or not:
SELECT * FROM pg_am WHERE amname='chemindex';
SELECT * FROM pg_am WHERE amname='sortedchemindex';Indextype chemindex and sortedchemindex can be used in the following way:
CREATE INDEX index_name ON table_name USING chemindex(structure_column_name);
or
CREATE INDEX index_name ON table_name USING sortedchemindex(structure_column_name);Example:
CREATE INDEX ttest_idx ON ttest USING chemindex(mol);
or
CREATE INDEX ttest_idx ON ttest USING sortedchemindex(mol);What is the difference between chemindex and sortedchemindex ?
-
substructure searching in tables where sortedchemindex is applied gives back the hits quickly in relevance-sorted order, that is hits that are most similar to the query molecule are returned first
-
similarity searching in tables where sortedchemindex is applied is also much quicker, especially when narrow similarity threshold is defined
-
indexing time is longer (approximately 1.7 times) in case of sortedchemindex
-
returning all hits in searches can be faster if chemindex was applied
{warning} Do not create indexes of both indextype on the same column simultaneously because it would increase the search time in the given table.
{warning} Neither indextype can handle NULL value. That means that columns containing NULL values can't be indexed and NULL value can't be added to an indexed column.
{info} Using the index in any type of searches can be forced by setting enable_seqscan parameter to OFF value in postgres configuration file
/etc/postgresql/9.5/main/postgresql.conf.{primary} We suggest closing all transactions and running a VACUUM before creating an index to avoid the calculation of the index data on rows that are being changed in the current transaction. E.g. if a column was added in the current transaction, the index creation time can be almost double of the normal time.
The same logic applies to updating a large number of molecules in an indexed table and then search on the table without closing the transaction and clearing up modification with VACUUM. If possible, update operations and column additions are advised to be performed before adding the chemical index.
Be aware that an ALTERed, not VACUUMed table can contain NULL values in the background and may force the index creation to stop.
MOLECULE IMPORT
All well known chemical structural data file formats are supported, but we provide functions for importing mol, sdf files.
{primary} In case of big files, the postgresql client can run out of memory as the following error message displays:
ERROR: out of memory DETAIL: Cannot enlarge string buffer containing 0 bytes by 1506367917 more bytesPlease, split up the original input file into smaller pieces and import them one after another. (Here is a script tool provided for splitting SD files.)
{primary} For importing purposes, usage of scripts consisting of multiple SQL insert statements is not recommended as this operation may be time consuming (especially if the destination table contains a chemical index). To speed up import, it is advisable to use the standard ‘copy’ SQL function or the
parse_sdfChemAxon utility.
Import from SD file
Import from local SD/mol files containing added fields besides the chemical structure data
The content of all the added fields in the SD file will be stored in a hashmap like type column.Optionally selected added fields from the SD file can be stored in separate columns as well.
Follow the next steps:
-
Set the content of the SD file to a variable (e. g.: content )
et variable_name `cat sdf_file_name`Example:
et content `cat ~/a.sdf`You can check the table generated by the
parse_sdffunctionSELECT * FROM parse_sdf(:'content');The
parse_sdffunction returns the content of the sdf file as a table, which has two columns:molSrc = the full sdf source of the original entry in the sdf file
props = hstore type column, which contains the properties of the sdf entry
You can also create a table similar to the table created by the
parse_sdffunction by:CREATE TABLE tableName (mol MOLECULE('sample'), p hstore); -
Create a table with
create table as:CREATE TABLE table_name AS SELECT molSrc::molecule('molecule_type_name') AS structure_column_name[, props ->'sdf_field_name' AS column_name] FROM parse_sdf(:'');Example:
CREATE TABLE mysdftable AS SELECT molSrc::molecule('sample') AS mol, props -> 'MOLFORMULA' AS formula, CAST(props -> 'CDID' AS INTEGER) AS id FROM parse_sdf(:'content');where
mysdftable = the name of the table
mol = the name of the column for storing structural data
molecule('sample') = the type of column mol
sample = molecule type (must be a defined type in
/etc/chemaxon/types/folder )MOLFORMULA = the name of the field in the SD file for storing chemical formula
formula = the name of the column for storing chemical formula
CDID = the name of the field in the SD file storing the identifier
id = the name of the column for storing the identifier integer
You can also store all properties from the SD file entry in a separate column in the structure table, and later, optionally, add additional columns to separately store selected properties.
Example:
et content `cat ~/a.sdf` CREATE TABLE mytable AS SELECT molSrc::molecule('sample') AS mol, props AS properties FROM parse_sdf(:'content'); ALTER TABLE mytable ADD COLUMN formula TEXT; UPDATE mytable SET formula = properties -> 'MOLFORMULA'
Available from version 1.8.
Running the steps below, the molecules in SD/mol files will be checked during the import and the invalid molecules will be stored in a separate table named <new_table_name>_error.
Steps:
-
Run the import_sdf.sql script to create function import_sdf . This script file is a customizable tool; you can update it according to your needs.
\i import_sdf.sql;{primary} Usage of function import_sdf is not recommended in case of tables already containing indexed chemical data as this operation may be time consuming. To speed up import, in this case a temporary table without indexes can be created with import_sdf and then the content of this temporary table can be inserted into the destination table using the following SQL statement:
insert into <destination table name> select (tt.mol, tt.props, ...) from <temporary table name> tt; -
Run the following commands including import_sdf function with the appropriate parameters:
et <variable_name> `cat <path_to_your_sdf_file>`; SELECT import_sdf(:'<variable_name>', '<new_table_name>', '<molecule_type>');Example:
et content `cat ~/a.sdf`; SELECT import_sdf(:'content', 'molecules', 'sample');
Import from (cx)smiles or (cx)smarts files
- Import from (cx)smiles or (cx)smarts files by the standard copy sql function *
COPY table_name (structure_column_name) FROM 'file_name' (FORMAT csv);Example:
COPY ttest(mol) FROM '/home/posgresuser/targetfiles/nci-pubchem_1m_unique.smiles' (FORMAT csv);Import from (cx)smiles or (cx)smarts files while collecting the invalid molecules
Available from version 1.8
Running the steps below, the molecules in smiles/cxsmiles/smarts/cxsmarts files will be checked during the import and the invalid molecules will be stored in a separate table named <new_table_name>_error . We propose the following two methods: PL/pgSQL script method and SQL commands method.
PL/pgSQL script method
We suggest applying this script method when the server and the client are on the same machine; or at least the molecule files are available on the server and their absolute path is known. Otherwise, apply the steps of SQL commands method.
Steps:
-
Run the import_single_line_format.sql script to create function import_single_line_format. This script file is a customizable tool; you can update it according to your needs.
\i import_single_line_format.sql; -
Run the following SELECT using import_single_line_format function with the appropriate parameters:
SELECT import_single_line_format('<absolute_path_to_my_file_on_server>', '<new_table_name>', '<molecule_type>');Example:
SELECT import_single_line_format('/home/myuser/molecules.smiles', 'molecules', 'sample'); -
Create a table which will contain the valid molecules
CREATE TABLE my_table(mol TEXT); -
Import from file
\COPY my_table FROM '~/molecules.smiles' (FORMAT csv); -
Create and load a table for the invalid molecule sources
CREATE TABLE my_table_error AS SELECT mol FROM my_table WHERE NOT is_valid_molecule(mol); -
Remove invalid molecules from my_table
DELETE FROM my_table WHERE mol IN (SELECT mol FROM my_table_error); -
Convert the valid molecule sources into molecule
ALTER TABLE my_table ALTER COLUMN mol TYPE molecule('sample') USING mol::molecule('sample');
Convert molecule source text to molecule from an existing table
Execute the 5th step of the SQL commands method above.
Cache loading
(Available form version 4.3.)
The cache is loaded when the first search is executed, but it can be loaded intentionally at any time using the following statements:
select load_molecule_cache('<index_name>');
select load_fingerprint_cache('index_name>');Both statements are recommended to be executed if the necessary memory is available.
In the case of low memory setup use only the load_fingerprint_cache function.
MOLECULE FUNCTIONS
Casting a string to any Molecule type defined in /etc/chemaxon/types/
'CCCC'::Molecule('sample'){primary} Postgres type system can cast a string to molecule if it is obvious; if the operation needs a molecule as an input. There are cases when there is no need of explicit casting because of the auto cast mechanism. See details here.
Example:
SELECT 'C' |<| 'CC'::molecule('sample');
Interpreting a string using a specific format
As molecule strings are ambiguous in some cases, it is possible to interpret the given molecule string according to the given molecule format using the molecule(String, String) function:
molecule(structure_string, molecule_format)where
structure_string = molecule string representation
molecule_format = molecule format string, see file formats
SELECT * FROM table_name WHERE molecule(structure_string, molecule_format) |<| column_name;A typical usecase is the differentiation between SMILES and SMARTS strings:
-
'CC' with SMILES notation is interpreted as two aromatic or aliphatic carbon atoms connected with a single bond.
-
'CC' with SMARTS notation is interpreted as two aliphatic carbon atoms connected with a single bond.
Example:
SELECT * FROM ttest WHERE molecule('CC', 'smiles') |<| mol;
SELECT * FROM ttest WHERE molecule('CC', 'smarts') |<| mol;Transformations
Transformation function is provided to transform the molecule structure according to specific needs. Multiple transformation strings can be provided separated by two dots.
query_transform(molecule, 'transformation_string1..transformation_string2')where
molecule = "molecule string" | Molecule object | table column
transformation string = "dbsmarkedonly" | "fullfragment"
Molecule transformation combined with substructure search:
SELECT * FROM ttest WHERE query_transform(molecule, 'transformation string') |<| mol;By default, CIS query matches only with CIS target and TRANS query matches only with TRANS target. If matching of CIS query with both CIS and TRANS targets or matching of TRANS query with both CIS and TRANS targets is aimed, then the query molecule has to be transformed.
The query_transform function with dbsmarkedonly option is provided to accomplish this transformation.
query_transform('query_structure', 'dbsmarkedonly')Examples:
SELECT * FROM ttest WHERE query_transform('C\C=C\C', 'dbsmarkedonly') |<| mol;
SELECT * FROM ttest WHERE query_transform('C\C=C\C', 'fullfragment..dbsmarkedonly') |<| mol;Full fragment search can be executed by transforming the query structure in a way that it matches only a full fragment of the target structure and by executing a substructure search on this modified query structure. The transformation adds 's*' query search property to all atoms.
query_transform(query_structure, 'fullfragment')Examples:
SELECT * FROM ttest WHERE query_transform('C1CCCCC1', 'fullfragment') |<| mol;Chemical terms
(Available since version 1.4.)
Calculation of chemical terms
Function chemterm makes possible to calculate chemical terms.
{primary} You may need to purchase separate licenses to apply function chemterm depending on the Chemical Terms used.
chemterm('chemical_term','structure')where
structure = a molecule string
chemical_term = a chemical term function
The output of function chemterm is one of the following formats:
-
string
-
molecule string in format mrv
Examples:
SELECT chemterm('name','CCO');
SELECT chemterm('mass()','CCO');
SELECT molconvert(chemterm('canonicalTautomer()','CC(O)=C')::Molecule,'smiles');Addition of chemical term columns
Chemical term columns can be added by using triggers.
See the following code block as an example:
CREATE TABLE test(structure MOLECULE('sample'), molweight NUMERIC);
CREATE OR REPLACE FUNCTION set_molweight()
RETURNS trigger AS
$BODY$
BEGIN
IF NEW.structure is null THEN
NEW.molweight:=NULL;
END IF;
NEW.molweight:=chemterm('mass()',NEW.structure)::real;
RETURN NEW;
END;
$BODY$
LANGUAGE plpgsql;
DROP TRIGGER IF EXISTS tr_molweight ON test;
CREATE TRIGGER tr_molweight BEFORE INSERT OR UPDATE ON test
FOR EACH ROW EXECUTE PROCEDURE set_molweight();
UPDATE test SET molweight=chemterm('mass()',structure)::real;Calculating chemical terms on the standardized structure
Chemical term is calculated on the input structure without standardization. Combine with the standardize method to calculate chemical terms on the standardized structure:
-
Standardize the structure before inserting into the database columns:
INSERT INTO <your_table> (mol, ... ) VALUES (standardize(<your structure>::Molecule('sample')), ... ) -
Store both the not standardized and the standardized structures:
INSERT INTO <your_table> (mol,standardized_mol ... ) VALUES (<your structure>::Molecule('sample'), standardize(<your structure>::Molecule('sample')), ... ) -
Standardize on the fly before calling chemical terms:
select chemterm(<your_chemical_terms>, standardize(<your_structure>::Molecule('sample'))::Molecule);
Standardize
(Available since version 4.4.)
Calculating the standardized structure
The standardization steps are determined by the molecule type definition. The returned value is a the standardized structure in MRV format
standardize(molecule)where
molecule = a Molecule object
Example:
SELECT standardize('C1=CC=CC=C1'::Molecule('sample'));Molconvert
Conversion to molecule formats
The use of ChemAxon's MolConverter is supported with some limitations:
molconvert('structure', 'format')where
structure = a Molecule in any of the following formats
format = mrv, mol, rgf, sdf, rdf, csmol, csrgf, cssdf, csrdf, cml, smiles, cxsmiles, abbrevgroup, sybyl, mol2, pdb, xyz, inchi, or name
Example:
SELECT molconvert('CC','sdf');Conversion to base64 encoded binary formats (image)
Molecules can be converted to binary image formats (png, jpeg, msbmp, pov, svg, emf, tiff, eps) or other binary formats (pdf) in Base64 encoded form.
Example:
SELECT molconvert('CC','base64:png');Molecule validation
Available from version 1.8.
The following function is provided to check the validity of the molecules:
is_valid_molecule(structure_text)where
structure _text = a Molecule in any of the following formats:
mrv, mol, rgf, sdf, rdf, csmol, csrgf, cssdf, csrdf, cml, smiles, cxsmiles, abbrevgroup, sybyl, mol2, pdb, xyz, inchi, or name
Example:
SELECT is_valid_molecule('CCO');You can filter out the invalid structures from a table using the following SELECT statement:
SELECT structure_text from mytable where is_valid_molecule(structure_text) = 'f';Relevance
Function relevance(Molecule) gives back a numeric type value based on the atom counts and further topological features of the molecule. This function can be applied for ordering search results.
Example:
SELECT relevance('CCO');MOLECULE OPERATORS
The main features of the different search types are available in JChem Query Guide.
The molecule operators work also in tables without chemical index, however, chemical index makes the search operations much faster.
{warning} JDBC Caution
Please take into account the JDBC related recommendation when implement structure searches.
Substructure search
Substructure search is performed using the symmetrical sub-/super-structure search operator: |<|.
SELECT * FROM table_name WHERE query_structure |<| structure_column_name;
SELECT * FROM table_name WHERE structure_column_name |>| query_structure;
SELECT * FROM table_name WHERE query_mol |<| target_mol;
SELECT * FROM table_name WHERE target_mol |>| query_mol;where
query_mol = "molecule string" | Molecule object | table column
target_mol = "molecule string" | Molecule object | table column
{warning} Query features and Markush features are not supported on target_mol.
Examples:
SELECT '[#6]-[#6]' |<| 'CC'::Molecule('sample');
SELECT * FROM ttest WHERE 'c1ccccc1' |<| mol;
SELECT * FROM ttest WHERE 'testmol
Mrv0541 01211514572D
3 2 0 0 0 0 999 V2000
1.2375 -0.7145 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1.9520 -1.1270 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.6664 -0.7145 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
1 2 1 0 0 0 0
2 3 1 0 0 0 0
M END
' |<| mol;See also molecule functions about defining the query structure format.
{primary} In order to get the hits quickly and sorted by relevance, please, create index on the structure column using sortedchemidex indextype instead of chemindex indextype.
See section Relevance sorting for more information.
Use of prepared statements
In case you plan to apply prepared statements for substructure search, please, take into account the comments relating PostgreSQL Server Prepared Statements like "Server side prepared statements are planned only once by the server. This avoids the cost of replanning the query every time, but also means that the planner cannot take advantage of the particular parameter values used in a particular execution of the query. You should be cautious about enabling the use of server side prepared statements globally. "
Superstructure search
Superstructure search is performed using the sub-/super-structure search operator: |>|.
SELECT * FROM table_name WHERE query_structure |>| structure_column_name;
SELECT * FROM table_name WHERE structure_column_name |<| query_structure;
SELECT * FROM table_name WHERE query_mol |>| target_mol;
SELECT * FROM table_name WHERE target_mol |<| query_mol;where
query_mol = "molecule string" | Molecule object | table column
{warning} Query features and Markush features are not supported on query_mol.
target_mol = "molecule string" | Molecule object | table column
Example:
SELECT * FROM ttest WHERE 'CCC' |>| mol;Full fragment (exact fragment) search
Full fragment search can be executed by transforming the query structure in a way that it matches only a full fragment of the target structure and by executing a substructure search on this modified query structure.
See details in transformations for full fragment search.
SELECT * FROM table_name WHERE query_transform(query_structure, 'fullfragment') |<| structure_column_name;Example:
SELECT * FROM ttest WHERE query_transform('CC', 'fullfragment') |<| mol;Duplicate search
Duplicate search is performed using the |=| operator.
SELECT * FROM table_name WHERE query_structure |=| structure_column_name;
SELECT * FROM table_name WHERE structure_column_name |=| query_structure;Example:
SELECT * FROM ttest WHERE 'CCC' |=| mol;Tautomer search
You have to apply such a molecule type which has tautomer = GENERIC tautomer mode.
How is chemical matching of the query and the target executed in tautomer search? The generic tautomer - representing all theoretically possible tautomers - of the target is matched with the query structure itself. This method is applied in substructure search, full fragment search, duplicate, and superstructure search.
Limitations:
-
SMARTS atoms and SMARTS bonds in the query structures are not supported.
-
The use of bond lists in query structures may slow down the search.
Similarity search
There are some differences between the similarity scores of the new method and of the old methods of JPC. The new method works on the standardized chemical structures while the old methods do not use the standardized format, only the original chemical structures. The new method provides better performance than the old methods. None of them supports query features on the query molecules, but they handle their presence differently.
New method
Available from JPC version 2.5. This new method provides better performance than the old methods, and there is no need to add extra fingerprint column to the table.
You can select those molecules and their similarity value from a table whose similarity value relating the given query structure is greater (or smaller) than a given similarity threshold value.
For this purpose the sim_filter or the sim_order type can be applied as shown below in the examples. The use of the sim_filter type results unsorted output, while the use of the sim_order type results sorted output.
For retrieving the most (or less) similar target molecules sorted by their similarity values, the sim_order type must be applied and the LIMIT n condition can also be useful for better performance if only the most similar (or dissimilar) molecules are required.
SELECT field1, field2, structure_column_name |~| 'query_structure' FROM table_name
WHERE ('query_structure', similarity_value)::sim_filter operator structure_column_name;
SELECT field1, field2, structure_column_name |~| 'query_structure' FROM table_name
WHERE ('query_structure', similarity_value)::sim_order operator structure_column_name LIMIT n; where
similarity_value is the similarity threshold value, a number between 0 and 1
operator can be |<~| (meaning similarity value less than or equal) or |~>| (meaning similarity value greater than or equal)
Examples :
SELECT mol FROM moltable WHERE ('CCC', 0.8)::sim_filter |<~| mol;
SELECT mol, mol |~| 'CCC' FROM moltable WHERE ('CCC', 0.8)::sim_filter |<~| mol;
SELECT count(*) FROM moltable WHERE ('CCC', 0.8)::sim_filter |<~| mol;
SELECT mol, mol |~| 'CCC' FROM moltable WHERE ('CCC', 0.8)::sim_order |<~| mol LIMIT 20;
SELECT mol, mol |~| 'CCC' FROM moltable WHERE ('CCC', 0.2)::sim_order |~>| mol LIMIT 20;Limitations:
- Query structures with query features (like list atoms, query atoms, query bonds, ... ) are not supported.
Old methods
Reaction search
Available from version 2.3.
The following search types are supported not only for molecules but for reactions as well.
Connection handling during searching
After retrieving the desired hits, the SQL connection needs to be closed in order to close the search on the jchem-psql server side. You can achieve this by having autocommit switch on or by calling commit explicitly.
Combination of structure query AND structure/non-structure query
-
WHERE condition referring to more than one column containing chemical structure data is not supported.
-
WHERE condition referring to one structure column and one or more columns containing non-chemical structure data is supported.
Relevance sorting
Relevance sorting of substructure search results is provided by JPC. The new method sorts the hit molecules according to the similarity between them and the query structure. This new method is much faster than the old method which sorts by relevance function. There can be some difference between the order of the hit molecules retrieved by the two methods.
Relevance sorting by using order by molecule column
This simple method is introduced in version 2.6.
In order to get the results quickly and sorted by relevance, the column storing the chemical structures must be indexed with sortedchemindex.
For executing substructure search the use of ORDER BY <molecule column> is essential and the LIMIT n parameter is recommended.
Example (assuming table "test" has column "mol" of type Molecule):
CREATE INDEX test_idx on test using sortedchemindex(mol);
SELECT mol FROM test WHERE 'c1ccccc1' |<| mol ORDER BY mol LIMIT 100;{primary} ORDER BY <molecule column> is only available for substructure search. In case of other search types, error message is given.
{warning} Applying ORDER BY <molecule column> in searches containing joins of several constraints, the execution plan optimizer of PostgreSQL may decide to choose a plan where a final sorting step is involved instead of keeping the original sorting order available in sortedchemindex. This may take very long time because sorting requires very expensive comparisons. In this case we advise to force the planner to choose a plan without sort by executing the command
SET enable_sort=OFF;It is necessary because PostgreSQL does not take into account the comparator function's cost in the cost of sorting.
Relevance sorting by using relevance function
Relevance sorting by using relevance function is deprecated.
AUXILIARY FUNCTIONS
Debugging
Raising the log level of the psql-client for debugging purposes:
SET client_min_messages to debug;Performance log
You can log the performance of the current session as:
SELECT * FROM perf_out();You can clear the log as (available since version 1.4):
SELECT perf_reset();Performance tuning of combined queries
Available since version 1.4.
The performance of searching with combined queries - when chemical structure query condition is combined with a non-structure query condition - might be improved by executing the following calibration steps.
-
Prerequisite: the column structure_column_name in table table_name used for the calibration must be indexed using indextype chemindex or sortedchemindex.
-
Calibration of cost factors
select calibrate_cost_factors('table_name', 'structure_column_name', 'query_structure');where
query_structure is an optional parameter. If not specified, chlorobenzene (Clc1ccccc1) is used by default.
{info} The applied query_structure is an important key factor of the calibration. The best specified query_structure has almost the same (+/- 10%) estimated selectivity in the given table as the number of its search hits. We recommend the next explain statement to run in order to get the estimated selectivity. The number of rows given in the output of the next statement is the estimated selectivity.
explain select * from table_name where 'query_structure' |<| structure_column_name;{primary} In order to get better performance, you may need to increase the
default_statistics_targetvalue for PostgreSQL analyzer. Its default value 100 can be increased to maximum 10000. -
Application of the calibrated cost factors
Please follow the suggested solutions given in the output of the
select calibrate_cost_factorsstatement.Example up to version 2.6:
To set the values permanently, add the lines
chemaxon.cost_slope_factor = -5.14e-07
chemaxon.cost_intercept_factor = 0.03866
to the PostgreSQL configuration file (e.g.: /etc/postgresql/9.5/main/postgresql.conf).
To set values only for the current session, execute the following commands:
SET chemaxon.cost_slope_factor = -5.14e-07;
SET chemaxon.cost_intercept_factor = 0.03866;
Example from version 2.7:
To set the values permanently, add the lines
chemaxon.index_screen_factor = 0.00002
chemaxon.index_abas_factor = 0.00895
chemaxon.seq_screen_factor = 0.00076
chemaxon.seq_abas_factor = 0.26312
to the PostgreSQL configuration file (e.g.: /etc/postgresql/9.5/main/postgresql.conf).
To set values only for the current session, execute the following commands:
SET chemaxon.index_screen_factor = 0.00002
SET chemaxon.index_abas_factor = 0.00895
SET chemaxon.seq_screen_factor = 0.00076
SET chemaxon.seq_abas_factor = 0.26312